分析锂离子的传输
发布时间:2018-08-30 09:20:18浏览次数:1685次
锂离子的传输
锂离子的传输对于电解质和电极材料都有着十分重要的意义,尤其是固态电解质。作为固态电解质中的一种,Li10GeP2S12正是由于其较高的锂离子电导率(12 mS/cm)而受到追捧。利用DFT可以有效地计算锂离子在固体中的传输扩散,其中NEB(Nudged elastic band)和AIMD是两种比较常用的计算方法。
NEB可以用来计算过渡态(Transition state),也可以用来计算锂离子在体系中的扩散。以层状Si为例,锂离子在其表面的扩散可以有以下几种方式,分别对应单层(SL)Si和双层(DL)Si结构。扩散能磊越小,意味着锂离子越容易扩散。可以看出不同的扩散方式以及不同浓度下锂离子的扩散能磊各有不同。在实际计算中,通常先优化初始和末态的结构,然后利用vtst的脚本对中间结构进行差值。提醒读者需要在插完值后对中间结构进行检查。

另外一种常用的计算方法是AIMD。由于其基于DFT,所以力场函数(Force filed)不用外加设定,这相比于经典的MD要更加方便。然而,由于其计算量要比经典MD大很多,所以一般的AIMD只适用于比较小的体系(几百个原子)。基于AIMD,可以利用Einstein关系式计算锂离子的扩散系数,即
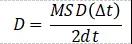
其中,MSD为锂离子的总平均自由移动距离,d为维数。
在实际应用中,对上式进行拟合时,需要特别注意起始段和结尾段。Mo等人针对Li7La3Zr2O12, Li10GeP2S12,Li1.33Ti1.67Al0.33(PO4)3材料的计算,提出以下的判据:MSD<0.5a2及?t>0.7ttot的数据舍去。其中a为锂原子位间距,ttot为总的MD时长。
根据Nernst–Einstein关系式,电导率可以计算为
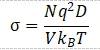
其中N为锂离子数,q为锂离子带电荷,V为体系体积,kB为Boltzmann常数,T为温度。一般的MD都是在高温下(~1000K)计算,然后经过Arrhenius关系式插值得到室温下的锂离子扩散系数,再进一步利用Nernst–Einstein关系式得到电导率。

注意这里只有扩散系数D对温度存在这样的函数,而则应考虑一起。

锂离子或小分子的吸附
做为正极材料,硫单质具有较高的比容量(1673mAh/g)。因而发展下一代可循环利用的商业锂硫电池已经提上日程。然而在反应中,锂离子和硫单质形成的多硫化锂溶于电解质并在正负极间穿梭,导致活性物质损失以及增加电解液的粘度。此外,溶解的多硫化物还会扩散到负极,与负极反应破坏电解质界面膜(SEI),增大界面电阻。因此在实际应用中,有效的限制多硫化锂的穿梭,使其集中在正极侧是一种有效的方法。
N掺杂石墨烯可以有效地抑制多硫化物的脱附,从而控制穿梭效应。Cheng等人研究了不同N掺杂结构石墨烯对多硫化物的吸附能,并得出结论认为石墨烯掺杂吡啶型聚类N可以有效地吸附多硫化物,如下图左侧所示。

在实际计算中,吸附能定义如下

其中对应于Li2Sx在基底(石墨烯)上吸附之后的结构的,是单独优化基地得到的能量,是多硫化锂小分子的能量。
在实际计算中,考虑到S原子和C原子之间比较强的范德华作用力,一般需要在DFT的计算基础上引入范德华修正。Zhang等研究了考虑范德华修正和不考虑范德华修正下多硫化锂和石墨烯的作用,发现引入范德华修正后吸附能会提高,而且趋势也有所变化。在不考虑范德华修正的情况下,多硫化合物和石墨烯几乎没有作用,吸附能在0.1~0.3 eV。在vasp中,考虑范德华修正的相关参数为IVDW。
材料稳定性的计算
材料稳定性是锂离子电池能长时间运行的得力保证,同样也是各种材料可以在实验室合成的基础。由热力学第二定律可知,相对于组分来说,平均自由能是一个凸包(convex hull),如下图。其中坐落在convex hull线上的点对应稳定相,如下图中的红点;而落在convex hull线以上的点则不稳定,如下图中的蓝点。对于非稳定相,定义其距离convex hull线的垂直高度为energy above hull,即Ehull。该数值越大,则说明该相越不稳定。一般认为Ehull小于100 meV/atom属于亚稳定,而Ehull大于该阈值则认为不稳定。因此计算不同组分下的自由能,并构建convex hull可以成功预测材料的稳定性。
在实际计算中,可以先构造出不同组分的机构,然后利用DFT进行结构优化,得到吉布斯自由能G。以平均到每个原子的自由能为标准,是各个组分的函数。以Li-Fe-P-O为例,构建好的convex hull投影到二维平面中就是常见的三角相图。

Ong等对LiFePO4的相图进行研究,并在此基础上发展出pymatgen软件包。该软件包基于Python并结合Material Project数据库,可以很方便的进行一系列高通量计算。
结论
第一性原理在锂电相关领域里面发挥着很重要的作用。利用DFT计算,可以有效地预测材料的电子性质,如带宽\费米能级,HUMO和LUMO等重要特性。此外,结合DFT和AIMD,可以有效的预测锂离子在材料中的传输及扩散,从而预测其锂离子电导率。最后,结合已有的材料数据库和相关开发包,可以方便的计算材料的稳定性。
标题:分析锂离子的传输 地址:http://www.batthr.com/news/hangqing/183421.html

咨询热线:400-9028-806020-89859916
客服QQ:3431741396(个人)3424946377(企业)
传真:020-38892397
客服邮箱:3431741396@qq.com

官方微信
粤ICP备12041652号 粤B2-20181492人力资源许可证
![]() 粤公网安备 44010602006150号
粤公网安备 44010602006150号
版权所有:广州中缆信息科技有限公司
[本站之人才及招聘,未经授权不得转载,否则追究其法律责任]

